

Myelodysplastic syndromes (MDS) is a clonal disorder of hematopoietic stem cells (HSCs) characterized by clonal hematopoietic stem cells (HSCs) with cytopenia, morphological abnormalities, genetic alteration, ineffective normal hematopoiesis, and frequent progression to AML. It has long remained unresolved how MDS cells, which are less proliferative, inhibit normal hematopoiesis and eventually come to dominate the bone marrow space. Despite several studies of mesenchymal stem cells (MSCs), one of the principal components of HSC niche supporting normal hematopoiesis, the molecular mechanisms underlying this process remain unclear. In this study, we examined the mechanism by which less-proliferative MDS cells outcompete normal hematopoiesis through the effects on MSCs using serially transplantable Abcg2-induced MDS/AML model we recently generated. The recipient-derived normal BM cells displayed a considerably lower colony output with markedly decreased numbers of the hematopoietic stem progenitor cells (HSPCs) . However, there were no direct effects on the colony-forming ability of the recipient HSPCs co-cultured with MDS/AML cells, indicating that MDS/AML cells inhibited hematopoiesis through alteration of bone marrow microenvironment, such as MSCs, rather than direct interaction between normal and malignant HSCs.
We next analyzed histological features of BM specimens. Interestingly, bone sections from the MDS/AML mice showed a reduced trabecular bone and narrowed growth plates. Moreover, micro computed tomography (micro-CT) analysis of the femora showed a significant reduction of the trabecular bone volume in the recipient mice transplanted with the MDS/AML BM cells. We detected decreased bone formation based on the calcein double labeling, but unchanged numbers of the TRAP-positive mononuclear or multinucleated (osteoclastic) cells in the MDS/AML samples, suggesting that the reduced bone volume was caused by suppressed bone formation. The impaired bone formation was also observed in the human MDS patients in terms of lower bone volume and decreased expression of BGLAP, one of osteogenic markers. In line with the above findings, single cell qRT-PCR analyses of mouse MSCs displayed downregulation of a line of osteolineage markers, indicating that MDS/AML cells suppress bone formation through inhibiting osteolineage differentiation of MSCs. Based on the findings, we next examined if re-induction of osteolineage differentiation of the MDS/AML-derived MSCs could rescue the potential of MSCs to support normal hematopoiesis. Importantly, the number of colony-forming cells (CFCs) was significantly restored by inducing differentiation of MDS/AML-derived MSCs toward osteogenic lineage both in vitro and in vivo. These results indicate that the impairment of osteolineage differentiation is the principal cause for an impaired normal hematopoiesis in MDS/AML, and that restoring the supportive niche will be a potential therapeutic option.
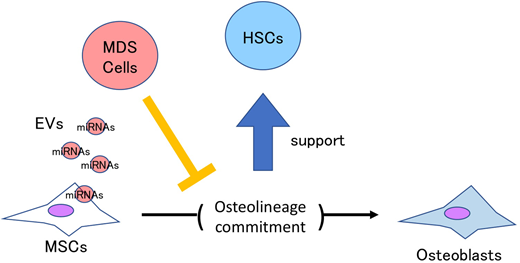
Since extracellular vesicles (EVs) derived from MDS/AML cells are critical mediators of intercellular communication, we examined the molecular mechanism underlying the dysfunction of MSCs via EVs. As expected, EVs from MDS/AML cells were incorporated into the normal MSCs where osteolineage marker genes were clearly downregulated, and the number of CFCs significantly decreased in the HSPCs co-cultured with MSCs treated by the MDS/AML-derived EVs. Moreover, by comprehensively analyzing microRNAs (miRNAs) enriched in EVs derived from MDS/AML cells, we identified several miRNAs that impaired the differentiation of normal MSCs. These results suggested that miRNAs in EVs derived from MDS/AML cells disrupted the hematopoietic supporting niche through suppressing an osteolineage differentiation of MSCs.
Here we uncover a heretofore unrecognized mechanism of bone marrow failure in MDS via the impairment of osteolineage differentiation in MSCs. EVs from MDS cells will be an attractive therapeutic target to restore the supportive niches, such as MSCs, for the remaining normal HSCs.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal